

Molecular geometry modeling is a powerful tool for understanding the intricate relationships between molecular structure and biological activity – a field known as structure-activity relationships (SAR). The main premise of SAR is that the biological activity of a molecule is dictated by its specific chemical structure, not only the connections between nuclei but also how the molecule is twisted and arranged in a three-dimensional configuration. The holy grail in SAR is to be able to predict how molecular configurations influence vital processes such as drug interactions, chemical reactivity, and protein functionality. If this were possible, scientists could predict the efficacy of a drug, as well as its side effects and toxicity, long before it is ever tested on people.
The vector-scalar interactive graph neural network (ViSNet) framework, developed by Microsoft, is a novel approach to molecular geometry modeling. ViSNet is designed to help researchers predict molecular properties, simulate molecular dynamics, and gain a more precise understanding of structure-activity relationships. As a result, ViSNet has the potential to help transform drug discovery, materials science, and other critical fields.
Our research aims to improve the interpretability of molecular data, reduce computing costs, and evaluate real-world application utility. “Enhancing geometric representations for molecules with equivariant vector-scalar interactive message passing” was published in Nature Communications (opens in new tab) in January 2024 and selected for “Editors’ Highlights” in both the “AI and machine learning (opens in new tab)” and “biotechnology and method (opens in new tab)” categories.
Spotlight: AI-POWERED EXPERIENCE

Microsoft research copilot experience
Discover more about research at Microsoft through our AI-powered experience
Geometry deep learning and SAR
Geometry deep learning is a method at the forefront of SAR investigations: a powerful computational approach that harnesses the power of deep-learning techniques to analyze and understand the three-dimensional structures of molecules. Traditional deep-learning methods primarily focus on processing data organized in grid-like structures, such as images or sequences of text. However, molecules are inherently three-dimensional entities with complex geometries, making them challenging to analyze using conventional deep-learning approaches. Geometry deep learning addresses this challenge by building specialized architectures and algorithms capable of handling three-dimensional data. These methods enable computers to learn and extract meaningful features from the spatial arrangement of atoms within molecules, capturing crucial information about their structure and behavior.
Despite significant recent strides in geometry deep learning, however, challenges persist. These include:
- Insufficient molecular interpretability – We are limited in our ability to understand and interpret the inner workings of deep neural networks when applied to molecular geometry modeling. While these networks excel at making predictions based on large datasets and complex patterns, they often operate as “black boxes,” meaning the rationale behind their predictions isn’t always understandable or transparent. In the context of molecular geometry, this lack of interpretability poses challenges in comprehending why certain molecular structures lead to specific outcomes, such as biological activity or chemical reactivity.
- Rapidly increasing computing costs as molecular size increases – As molecules increase in size and complexity, the computational resources required to analyze them escalate dramatically. This challenge becomes particularly pronounced when employing advanced computational techniques, such as those using high-order Clebsch–Gordan coefficients. The Clebsch–Gordan coefficients are mathematical quantities used in quantum mechanics to describe the coupling of the angular momentum properties of particles. In the context of molecular modeling, these coefficients are employed in sophisticated quantum mechanical calculations to help account for the interactions between electrons and nuclei within a molecule. For large molecules, the number of atoms and electrons involved increases exponentially, resulting in an astronomical number of possible interactions that must be considered. As a result, the calculations involving high-order Clebsch–Gordan coefficients become tremendously complex and computationally demanding.
- Need for blind tests and evaluations in real applications – Assessing predictive models in real-world applications through blind tests is crucial for evaluating their reliability and applicability beyond controlled benchmarks. However, challenges arise due to the scarcity of diverse and representative datasets, and complex system dynamics. There are also ethical considerations in animal and human trials, which naturally restrict the availability of such data. Overcoming these challenges requires interdisciplinary collaboration, innovative methodologies, and transparent validation frameworks to ensure the robustness and trustworthiness of predictive models in addressing real-world challenges.
Enhancing molecular geometry representations by ViSNet
Originally, our goal was to develop a model capable of effectively harnessing the intricate structures of molecules. Traditional molecular dynamics (MD) simulations track molecular movements by considering factors like bond length, bond angle, and dihedral angles. Taking inspiration from these methods, we introduced a novel approach called the vector-scalar interactive graph neural network (ViSNet).
Instead of directly integrating bond angle and dihedral information into our model in a straightforward manner, we introduced a concept termed “direction units.” These units represent nodes within the molecular structure as vectors, calculated by summing normalized vectors pointing from the central node to its neighboring nodes. We expanded traditional calculations of bond length, bond angle, and dihedral angles into interactions involving pairs of atoms (two-body), triplets of atoms (three-body), and quadruplets of atoms (four-body). To efficiently manage these interactions, we devised a runtime geometry calculation (RGC) module, which accurately captures the complex relationships between atoms in a molecule. Remarkably, the RGC module’s computations for three-body and four-body interactions exhibit linear time complexity, ensuring computational efficiency.
Additionally, we introduced a mechanism known as vector-scalar interactive message passing (ViS-MP), facilitating the exchange of information between nodes and edges in the molecular graph. This mechanism iteratively updates the direction units of nodes based on scalar representations of nodes and edges, and vice versa, through the RGC module. These distinctive features of the RGC and ViS-MP significantly enhance our model’s capacity to encode molecular geometry and streamline the process of information exchange within the molecular graph neural network.
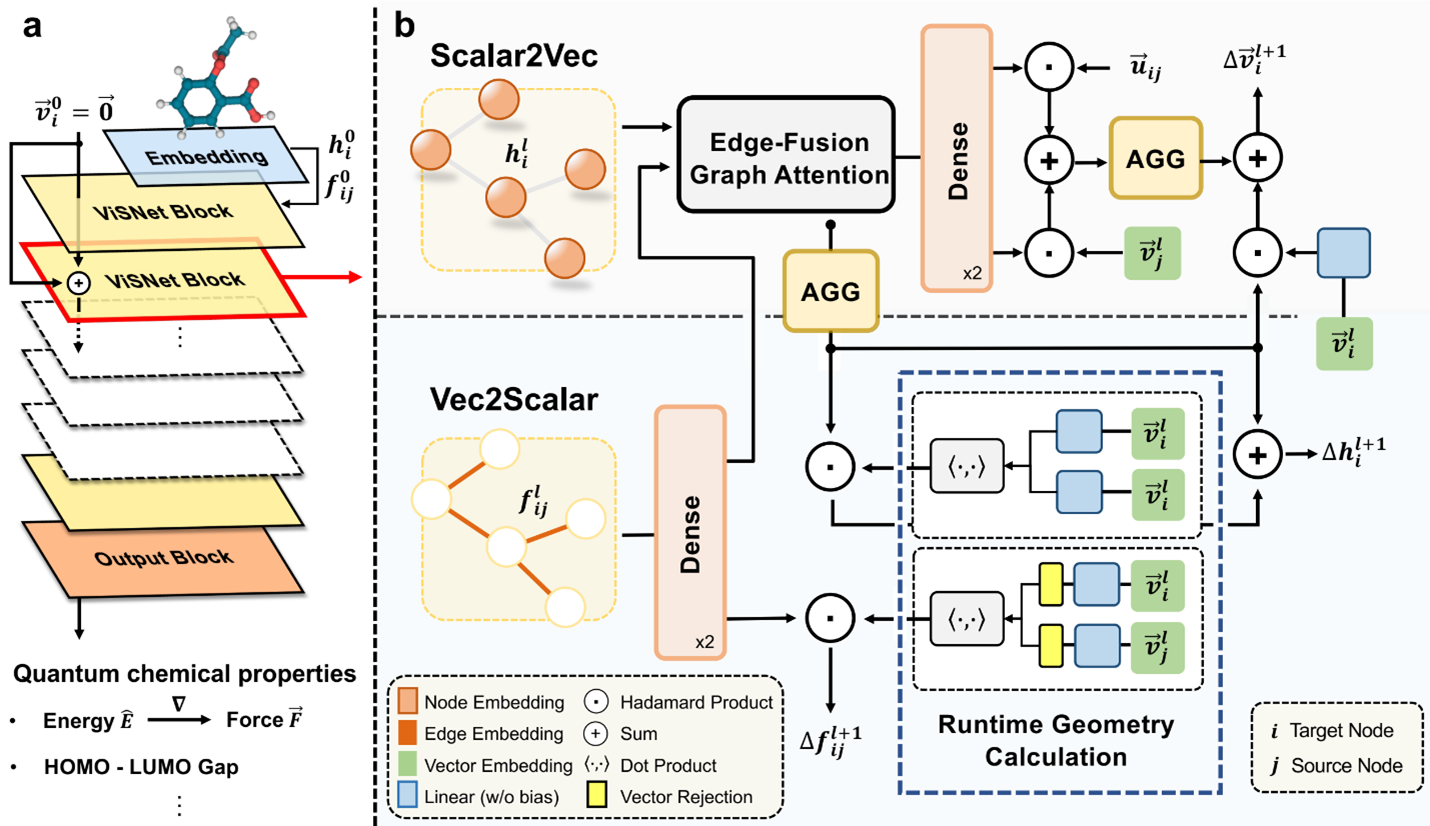
ViSNet in real-world applications for molecular modeling and property predictions
To gauge ViSNet’s practical utility, we rigorously evaluated its performance using established benchmarks for predicting molecular properties. Across a range of datasets, including MD17, revised MD17, MD22, QM9, and Molecule3D, ViSNet consistently outperformed existing algorithms, showcasing its exceptional accuracy in representing molecular geometry.
We then put ViSNet to the test by simulating the behavior of the Chignolin protein through molecular dynamics (MD) simulations. Trained on the AIMD-Chig dataset, featuring protein data calculated using advanced density functional theory (DFT) methods, ViSNet outperformed traditional empirical force fields and showed promise when compared to contemporary machine-learning force fields. Notably, simulations with ViSNet closely mirrored outcomes from rigorous DFT calculations, highlighting its potential for precise and efficient data simulations.
We used ViSNet to participate in the First Global AI Drug Development Competition (opens in new tab), an international competition to predict the inhibitors against the main protease of SARS-CoV-2, given the sequence information (i.e., SMILES) of small molecules. Worldwide, 1,105 participants from 878 teams took part in the competition. ViSNet helped us win the competition, demonstrating its promising prediction accuracy.
To make ViSNet more accessible and user-friendly, Microsoft has integrated it into the PyTorch Geometric Library (opens in new tab) as a core model for molecular modeling and property prediction. This integration aims to broaden the scope of applications and simplify the usage of ViSNet for researchers and practitioners. Additionally, to ensure ongoing support and improvement, a regularly updated version of ViSNet is now available on GitHub (opens in new tab), providing users with the latest enhancements.
Recognizing the potential limitations of graph neural networks, such as the risk of “over-smoothing” (i.e., making nodes indistinguishable from one another) as models grow larger and more complex, we developed a Transformer-based version of ViSNet known as Geoformer (short for Geometric Transformer). This novel variant, introduced in our publication at NeurIPS 2023 (opens in new tab), addresses scalability challenges by transferring the key components of ViSNet into the Transformer architecture. This includes incorporating the RGC module into the Transformer attention mechanism and introducing a new method called interatomic positional encoding (IPE) to capture spatial relationships between atoms.
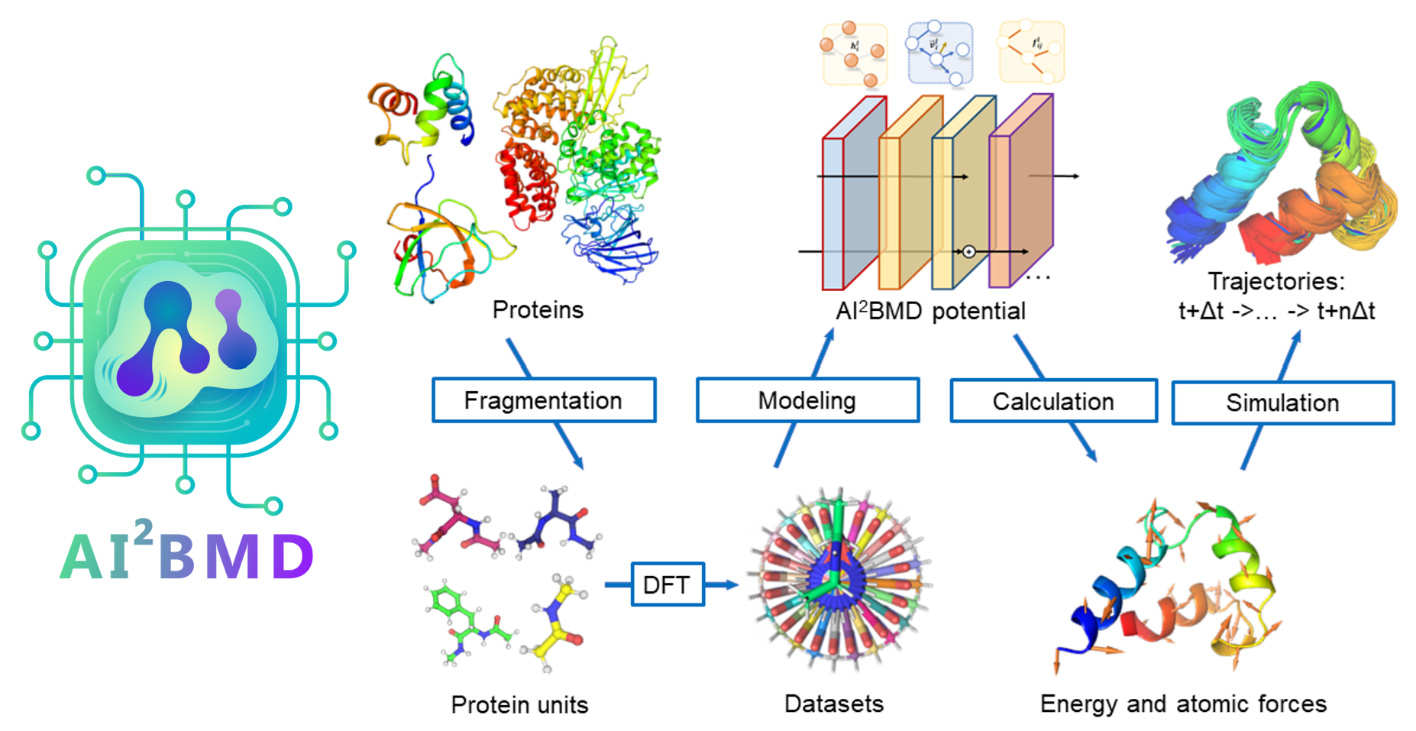
Looking forward: Toward AI-powered MD simulations with ab initio accuracy
As a crucial component of the AI-powered Ab Initio Molecular Dynamics (AI2BMD) project (opens in new tab), ViSNet plays a pivotal role in accelerating molecular dynamics simulations. The project’s primary objective is to enhance the accuracy and efficiency of these simulations, with the aim of achieving results comparable to those obtained through rigorous ab initio methods, even for large molecular systems.
By integrating ViSNet into AI2BMD, significant strides have been made toward achieving this goal. ViSNet enables AI2BMD to achieve levels of accuracy in energy and force calculations that closely approach those of ab initio methods, even for complex proteins containing over 10,000 atoms. By leveraging ViSNet in protein dynamics simulations, AI2BMD aims to enhance the precision of free energy estimations and provide valuable insights into protein folding thermodynamics.
ViSNet’s contributions extend beyond energy calculations to the characterization of various protein properties. These insights have the potential to complement experimental research efforts by offering predictive capabilities and guiding further investigations into protein structure and function. The advancements in molecular geometry modeling, demonstrated by the innovative ViSNet framework, portend a new era of precision and efficiency in computational chemistry and biophysics.
Through meticulous design and rigorous validation, ViSNet has emerged as a versatile tool capable of giving insight into the intricate relationships between molecular structure and biological activity – getting us one step closer to the holy grail of structure-activity relationships. The integration of ViSNet into established libraries and frameworks, coupled with ongoing research efforts to enhance scalability and accuracy, underscores its potential to revolutionize drug discovery, materials science, and more.