PhyloD
Machine learning tools for modeling viral adaptation to host immune responses.
 Research
Research
Nature Medicine | , Vol 22: pp. 606-613
Bonnie Mathieson Young Investigator Award
Additional links:
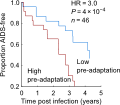 HIV adapts to our immune response. So what? That’s been a surprisingly difficult question to answer, beyond very focused questions about very special epitopes. So we teamed up with Paul Goepfert (opens in new tab) of UAB, Eric Hunter (opens in new tab) of Emory, and a several other labs to answer the question. First, we built a model of HIV adaptation and trained it on 4000 people. Armed with this model, we looked at a number of data sets to see how adaptation predicts disease progression, then Paul’s designed a series of functional studies to validate the results. Not only does HIV adaptation within a patient predict rapid disease progression, but infection by a pre-adapted virus–a virus that already carries mutations specific to the new host–results in dysfunctional immune responses and rapid progression. Thus suggests HIV is finding universal holes in our immune response, and bolsters claims that we should be pursuing vaccines that target regions of the virus that are relatively conserved. Moreover, these results highlight the interactions between host and virus genetics, explaining many of the “protective” effects commonly attributed to HLA alleles, and confounding estimates such as the “heritability” of viral load that ignore such interactions.
HIV adapts to our immune response. So what? That’s been a surprisingly difficult question to answer, beyond very focused questions about very special epitopes. So we teamed up with Paul Goepfert (opens in new tab) of UAB, Eric Hunter (opens in new tab) of Emory, and a several other labs to answer the question. First, we built a model of HIV adaptation and trained it on 4000 people. Armed with this model, we looked at a number of data sets to see how adaptation predicts disease progression, then Paul’s designed a series of functional studies to validate the results. Not only does HIV adaptation within a patient predict rapid disease progression, but infection by a pre-adapted virus–a virus that already carries mutations specific to the new host–results in dysfunctional immune responses and rapid progression. Thus suggests HIV is finding universal holes in our immune response, and bolsters claims that we should be pursuing vaccines that target regions of the virus that are relatively conserved. Moreover, these results highlight the interactions between host and virus genetics, explaining many of the “protective” effects commonly attributed to HLA alleles, and confounding estimates such as the “heritability” of viral load that ignore such interactions.
Human leukocyte antigen class I (HLA)-restricted CD8+ T lymphocyte (CTL) responses are crucial to HIV-1 control. Although HIV can evade these responses, the longer-term impact of viral escape mutants remains unclear, as these variants can also reduce intrinsic viral fitness. To address this, we here developed a metric to determine the degree of HIV adaptation to an HLA profile. We demonstrate that transmission of viruses that are pre-adapted to the HLA molecules expressed in the recipient is associated with impaired immunogenicity, elevated viral load and accelerated CD4+ T cell decline. Furthermore, the extent of pre-adaptation among circulating viruses explains much of the variation in outcomes attributed to the expression of certain HLA alleles. Thus, viral pre-adaptation exploits ‘holes’ in the immune response. Accounting for these holes may be key for vaccine strategies seeking to elicit functional responses from viral variants, and to HIV cure strategies that require broad CTL responses to achieve successful eradication of HIV reservoirs.
HIV is characterized by a tremendous rate of mutation, which leads to a high level of genetic diversity within and among patients. This genetic variation is the substrate for rapid within-host evolution. So as our immune system learns to target the virus, the virus adapts, leading to an endless game of cat-and-mouse. From a scientific perspective, this provides a useful opportunity: if HIV is contantly adapting to our individual immune responses, then studying HIV adaptation will provide insights into both virus function and basic immunology. Over the years, we have developed models of viral adaptation that have allowed us to do just that. Through these studies, we’ve learned that the cellular immune response (driven by CD8+ T-cells) is the primary driver of HIV within host adaptation. This arm of the immune system is dedicated to identifying cells that have gone haywire–for example, because of viral infection–then eliminating those cells before they cause problems. Not only do these responses drive HIV evolution they also (not coincidentally) appear to be central in determining rate of disease progression a person will experience. Interestingly, the details of this response depend on each individual’s genetics. Specifically, the genetic variations a person as at the HLA-A, HLA-B and HLA-C genes. These genes encode proteins that constantly sample the space of protein fragments (called “epitopes”) present in the cell and presents some of the on the cell’s surface so the CD8+ T cells can surveil them. There are thousands of variations of these HLA alleles, and each one presents slightly different epitopes. See our most recent review for more details.
But what is the clinical impact of all this adaptation? Does the immune system just keep on adapting itself, or does it eventually run out of options? And if it does run out of options, what happens if a person is infected with a “pre-adapted” virus–that is, one that’s already carrying a lot of the escape mutations that are specific to the person’s HLA alleles? Remarkably little is known about this. Yet there is a lot riding on opposing assumptions about the answers. For example, HIV appears (opens in new tab) to be slowly adapting to the HLA alleles present in different geographical regions. Is this a problem? If being infected by a pre-adapted virus is bad, then this is a really big problem! But then, if it’s not a problem, then we should be designing vaccines that target these variants, as is currently be pursued (opens in new tab).
To reach some clarity on this subject, we developed a new machine learning algorithm that builds on our previous models. In short, our goal was to estimate how likely it is that you would see a particular HIV sequence in a particular subject, as determined by the subject’s HLA alleles. If very likely, then we say the virus is well Adapted to the individual; otherwise, it’s not. If you’re curious, the underlying machinary is based on extensions of the phylogenetic dependency network (opens in new tab) we introduced years ago. We then teamed up with a dozen different labs who had collected HIV sequence and HLA types from a combined ~4000 individuals, so that we could train the models. Once we have trained models, we can ask the very simple question, How adapted is this virus to that person? And more interestingly, does adaptation predict disease progression?
The results were even stronger than we expected: not only is adaptation of a person’s virus a strong predictor of their current disease status, as measured by viral load and CD4 counts, but it predicts future changes in these markers. But more surprisingly, individuals who had the bad luck of being infected by a pre-adapted virus progressed 3 times faster to low CD4 counts and had much higher viral loads (Figure 3 in the paper). These effects were stronger even that the specific HLA alleles a person has (which has been the strongest predictor of progression in dozens of studies), and we replicated these effects on multiple cohorts, from both Africa and North America.
So what does it all mean? Some high level implications:
You don’t want to be infected by who’s virus is already adapted to your genetics! Among other things, this means where you’re infected will predict clinical outcome, since different regions have different circulating strains (see our Figure 4).
This suggests that some of the epidemiological markers we like to look at are oversimplified. For example, we showed that the extent to which circulating viruses were adapted to individual HLA alleles largely explained why some alleles are protective and some are not. On the viral genetics side, the extent to which viral genetics “explains” differences in viral load is hugely dependent on adaptation–that is, an interaction between human and viral genetics (Figure 4). That suggests that large scale epidemiological sequencing efforts, such as the amazing Pangea project (opens in new tab), really need to be collecting human genetics if they’re interested in clinical outcomes. Really, any clinical trial that lists Viral Load or CD4 decline as an end point should be collecting both viral sequences and HLA types. Looking at only one (or neither) introduces a huge amount of variation in outcomes.
Functionally, this implies there are some universal holes in the immune response–variants that you simply cannot learn to recognize. Why is that (and is it related to TCR bias (opens in new tab))? Noteably, Victor Du and Paul Goepfert looked at functional data to show that responses to adapted epitopes are rarer, and even when present, are less likely to actually kill the virus (Figure 5).
For vaccine design, it suggests challenges for the “mosaic” vaccine approach, which specifically seeks to include adapted variants in the vaccine. If we can’t get functional responses to those epitopes in the context of natural infection, it will be quite the challenge to get super-natural responses induced by a vaccine. At the very least, we can measure how adapted vaccine inserts are to individuals (Supplementary Figure 10), so we should be doing that and relying on tedious and costly killing assays to determine if the responses a vaccine induces are actually worthwhile. A critical open question here is whether adding adapted variants to non-adapted variants in a vaccine insert will strengthen the response, have no effect, or weaken the response. Our results suggest at the least that the latter is a possible concern.
On the other hand, these results are a strong validation of the idea behind conserved elements vaccines, which specifically seek to elicit responses to epitopes that do not have many variants circulating globally. Though care should be taken that we aren’t targeting conserved regions that encode adapted variants!
This work was the result of a truly collaborative effort that has been building over many years. Training the model required data from 4000 subjects, which we took from roughly a dozen different cohorts. We added to that data from the Zambian transmission pairs cohort (opens in new tab) and the HVTN 502 Step Study (opens in new tab). Based on our statistical results, Paull Goepfert’s lab designed some beautiful studies to measure the efficacy of immune responses to founder viruses, recruiting patients from the Alabama Vaccine Research Clinic at UAB (opens in new tab).
The key groups that drove this work:
University of Alabama at Birmingham (opens in new tab)
Emory University (opens in new tab)
Oxford University (opens in new tab)
Simon Fraser University (opens in new tab)
International AIDS Vaccine Initiative (IAVI) (opens in new tab)
https://mix.office.com/watch/1xbw46b42skuy (opens in new tab)
Or view it directly from the Office Mix website (opens in new tab).
Machine learning tools for modeling viral adaptation to host immune responses.