
Jonathan M. Carlson
Managing Director, Health Futures
About
I am the Managing Director of Microsoft Research Health Futures (opens in new tab) , Microsoft’s multidisciplinary research organization with the mission to empower every person on the planet to get the right treatment at the right time. We are focused on catalyzing the emergence of multimodal biomedical generative AI that can reason from bedside to bench, accelerating the discovery, development and delivery of medicine. To this end, Health Futures’ portfolio includes deep technical investments in generative AI applied to the languages of both humans and biology, spanning medical text to images, and DNA to proteins, cells, and immune systems. We collaborate closely with external partners to ground our research and incubation in high-impact scenarios, and work across the Microsoft ecosystem to bring innovations to the world. As a computational biologist, I have collaborated with dozens of labs across the globe, publishing over a hundred papers at the intersection of machine learning, immunology and virology. I hold a Ph.D. in computer science from the University of Washington and an undergraduate degree in biology from Dartmouth College.
Previously, I led the Antigen Map Project, a partnership with Adaptive Biotechnologies (opens in new tab) that has led to multiple clinical diagnostics and scientific discoveries using ML-based decoding of immune system genetics. The basis of this approach is Adaptive’s immunosequencing technology, which provides high-throughput sequencing of T-cell receptors, effectively turning a blood sample into an encoded representation of a person’s immunological history. By decoding this information, we could in principle develop diagnostics for autoimmune diseases, infections and cancer, along with identifying targets for therapeutic and prophylactic vaccines and engineered T cells.
Before that, I was deeply involved in Project Premonition, where I led the metagenomics efforts: given a sample of unknown DNA, what organisms contributed the genetic material? In the context of Premonition, we get DNA from mosquitos. Such DNA will come from the mosquito, the host(s) on which it fed, the mosquito’s microbiome, and both vector-borne viruses and blood-borne viruses from the mosquito host.
Much of my early published research focused on using virus evolution as a window into the host immune response, with HIV serving as a particularly useful substrate. Because HIV has a high rate of mutation, each HIV-positive individual carries a genetically distinct virus. Moreover, as the adaptive immune response learns to target the virus, evolution selects for genetic variants that reduce the effectiveness of the immune response, leaving genetic “footprints” on the virus that we can learn to track. So by developing models of virus evolution, we can generate and test hypotheses about how the immune system interacts with the virus. In addition to providing guiding principles for vaccine design, this approach can reveal fundamental new insights into basic immunology. Some of those papers are featured below.
Publication Highlights
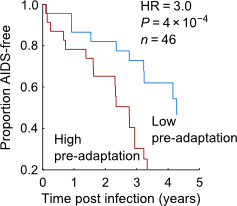
Impact of pre-adapted HIV transmission (Nature Medicine May 2016)
HIV adapts to our immune response. So what? That's been a surprisingly difficult question to answer, beyond very focused questions about very special epitopes. So we teamed up with Paul Goepfert of UAB, Eric Hunter of Emory, and a several other labs to answer the question. First, we built a model of HIV adaptation and trained it on 4000 people. Armed with this model, we looked at a number of data sets to see how adaptation predicts disease progression, then Paul's designed a series of functional studies to validate the results. Not only does HIV adaptation within a patient predict rapid disease progression, but infection by a pre-adapted virus--a virus that already carries mutations specific to the new host--results in dysfunctional immune responses and rapid progression. Thus suggests HIV is finding universal holes in our immune response, and bolsters claims that we should be pursuing vaccines that target regions of the virus that are relatively conserved. Moreover, these results highlight the interactions between host and virus genetics, explaining many of the "protective" effects commonly attributed to HLA alleles, and confounding estimates such as the "heritability" of viral load that ignore such interactions.
HIV-1 adaptation to HLA: a window into virus–host immune interactions (Trends in Microbiology April 2015)
This review with Zabrina Brumme gives an overview of HLA-mediated escape. We go over the history of HLA-mediated escape, showing how studying HIV adaptation has lead to fundamental insights into virology, immunology, and vaccine design. In effect, the rapid rate of HIV mutation, coupled with the astonishing plasticity of the virus, means that the virus is constantly exploring ways to adapt to its environment. Studying these adaptations provides an excellent starting point for understanding how the immune system works and what factors constrain viral evolution
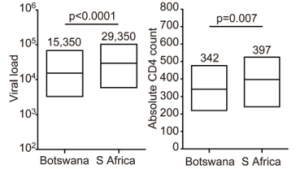
Is drug rollout reshaping pathogenesis? (PNAS December 2014)
Here's a provocative thought: as we roll out drugs to the sickest people first, are we selecting for weaker viruses--ie, those that don't make people sick, and thus are less likely to be subjected to drug therapy? We don't have direct evidence for this, but when we compare Botswana to South Africa, we see high CD4 (healthier immune systems) per level of viral load (viral concentration) or viral replicative capacity (how well it grows in a lab). Perhaps related (perhaps not), we also see an increased burden of circulating HLA escape mutations. At the very least, this increased burden appears to have wiped out B*57's ability to modulate relative viral control. Might it also have weakened the virus?
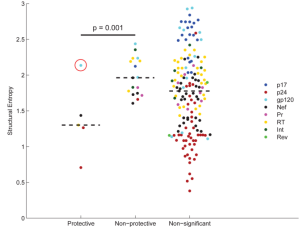
Natural controllers tend to target structurally constrained epitopes (Journal of Virology, November 2014)
This is a great paper that provides a great rationale for vaccine design: (1) it's critical to target specific epitopes; (2) those epitopes need to be those where mutation comes at a cost; and (3) protein structure is a great way to predict which epitopes will be costly. We did this in collaboration with Florencia Pereyra and Bruce Walker at the Ragon Institute. The idea was to test a bunch of nature controllers and normal non-controllers to see which epitopes they target, whether that explains control, and what characterizes good epitopes.
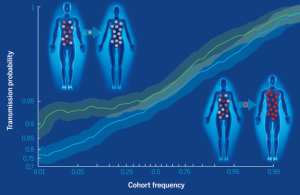
A fitness bottleneck in HIV transmission (Science, July 2014)
HIV is characterized by a tremendous rate of mutation, that leads to a high level of genetic diversity within and among patients. Yet transmission is frequently (~90%) established by a single genetic variant. What (if anything) is so special about that "founder" virus? In short, fitter viruses are more likely to be transmitted. This has two major implications: (1) there are likely many nonproductive infection events that happen at the site of exposure (otherwise, where is the substrate for competition?); (2) As you raise the bar for infect (that is, make it less likely you'll be infected), you increase the risk that breakthrough infection will cause more severe disease.
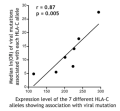
HLA-C expression and HIV immune control (Science, May 2013)
In collaboration with Mary Carrington's group, we showed in Science that the quantity of HLA-C surface protein correlates with HIV disease progression, the probability than HLA-C epitopes will be targeted by the immune system, and the probability that HIV will escape within those epitopes. Mary further showed that HLA-C expression levels are linked to some auto-immune diseases. This is an important study that highlights the the role of HLA-C (which is generally ignored in the field) and demonstrates how our models of selection can be used to generate and test hypotheses. As always, this was a hugely collaborative effort, making key use of data from Philip Goulder, Zabrina Brumme and many others.

Correlates of protection from HIV immune escape (Journal of Virology, December 2012)
As part of the IHAC collaboration, we published the largest HIV escape study ever done today. We studied the full HIV proteomes from 1,888 chronically HIV clade B-infected individuals who had never been given drugs to identify HLA escape mutations. This will be a useful resource to the community, and also showed some important new insights into HIV escape. For example, we now know that escape typically happen at anchor residues and that a hallmark of protective HLA alleles it the ability to drive escape across the proteome, especially at anchors.

Widespread impact of differential escape (Journal of Virology, January 2012)
This paper marks the development and introduction of our phylogenetically corrected logistic regression algorithm. This allows us to do all the standard logistic regression analyses--test for differential effects or measure effect size--as logistic regression, but do it while correcting for phylogenetic structure. You can use the tool yourself here, though we have to limit to single analyses. If you'd like an executable version of the code, email me. We're working on a better, scalable solution, so stay tuned. We used this approach to look at an interesting phenomenon: although we like to group HLA alleles by the their tendency to bind similar epitopes, we find that, in vivo, the escapes that evolution selects for differ by HLA. For example, when B*57:03 and B*57:02 (two very similar HLA alleles) present the same epitope, the observed escape mutations are usually different. Very surprising indeed, as it forces us to think more carefully about how (and if) we group alleles, as well as what the role of differential escape is. This work was in collaboration with Philip Goulder, John Frater, Roger Shapiro and Thumbi Ndung'u and thier labs.

HIV adapts to the innate immune response (Nature, August 2011)
Teaming up with Galit Alter and Marcus Altfeld from the Ragon Institute, we showed that HIV is adapting to the NK-cell-mediated immune response. We used PhyloD to identify HIV polymorphisms that are enriched among patients who express certain KIR genes. These associations imply that HIV is adapting to something specific in these individuals. In fact, NK-cells are activated and inhibited by their KIR proteins, which bind to HLA-epitope complexes. It looks like HIV mutates to manipulate these interactions, effectively shutting down the Natural Killer cells. What a great example of how we can start from adaptation, then work backward to figure out what's going on!

Phylogenetic dependency networks (Journal of Computational Biology, November 2008)
This paper is the introduction of the Phylogenetic Dependency Network framework. The idea is that we build independent models of evolution for each amino acid in an HIV protein. One of those models is parameterized by the phylogenetic structure, the rate of evolution in the absence of escape, and a model of adaptation in the leaves of the phylogeny. Crucially, we assume that adaptation exists only in the leaves (ie, the observed patients). This is clearly wrong, but quite useful in that it keeps the number of parameters linear, and empirically it's a decent approximation, as we showed previously. This paper is the foundation of all of our HIV escape work and has been cited numerous times. The image on the left was used as the cover image for this article, as well as the PLoS T-shirt logo for the 2009 ISMB conference!