
The drug development process is an iterative one that consists of discovery, design, and testing. Historically, drugs were derived from plants and discovered through trial-and-error experiments. Fortunately, this drug discovery process now occurs in a lab, with each iteration of custom-designed compounds producing a more promising candidate. While much safer and more effective, this process takes a great deal of time and money. In fact, it can take over 10 years to bring a single drug from the first stages of development to market and cost between $1–2 billion. A significant time investment occurs in the early stages—during the multiple cycles of designing and synthesizing new candidate molecules, testing them, and determining which molecular properties to improve before starting the cycle again. In fact, the steps of synthesis and in vitro testing of molecule behavior in a laboratory are inherently slow.
One way to speed up the drug-development process is through computational modelling so that most molecules can be prioritized in silico even without being physically available, and only the molecules most likely to succeed are synthesised and measured.
To enable such a speedup through computational modelling, a machine learning (ML) model must be able to precisely predict molecular properties, and in particular, whether a proposed drug molecule will be active — that is, able to affect the protein target associated with the disease.. ML is known to be very good at recognizing patterns in images and text, where millions of lines of such data are available. However, at the initial stages of the drug-discovery process, often only a few dozen molecules are likely to have been measured in a laboratory.
While little data is available for a drug discovery project from which an ML model can extract patterns, the data for tens of thousands of past projects is available in public and proprietary databases. Fortunately, we can use this data for molecular property prediction by having the ML model learn from the combination of these many related datasets.
- Event NeurIPS 2021
In collaboration with Novartis, the Machine Intelligence team at Microsoft Research Cambridge worked to address the problem of molecule-protein binding prediction given a limited amount of data. Our goal is to help both the ML and computational chemistry communities unite in making progress on this challenging problem. To this end, we’re publishing a paper at the thirty-fifth Conference on Neural Information Processing Systems (NeurIPS) 2021, FS-Mol: A Few-Shot Learning Dataset of Molecules.
In this work, we curated a few-shot learning dataset of molecules (opens in new tab) that reflects this challenge by providing small datasets for protein-ligand binding prediction alongside a principled method for using these datasets in few-shot learning. Such a dataset was not previously available, so we released an open-source evaluation framework that enables ML researchers to compare their work and helps drug discovery experts easily see which computational modelling approaches are most promising for their specific goals.
Applying few-shot learning to drug-discovery
Few-shot learning is a widely used concept in the computer vision and reinforcement learning communities. It entails the use of training data from a collection of associated tasks to prepare an ML model before adapting it to a new task of interest using only a few relevant datapoints. Much like how a human brain learns to recognise an object it has seen only once, the structure of the model is primed to pick up new information, so access to millions of datapoints for each new task we may encounter isn’t necessary. A few-shot learner is pretrained using an array of available datasets. The hope is that including a sufficiently large range of training tasks will ensure that at least some are similar to the eventual testing task of interest. In the drug discovery process, an example is the prediction of molecular binding to a specific protein. As shown in Figure 2, once pretraining has occurred, the few-shot learner is fine-tuned using a small amount of labelled training data (often termed the support set), which here consists of the small number of measurements made on synthesized molecules against the protein target. The resulting model is then evaluated by its ability to make predictions on held-out test data points (known as the query set).
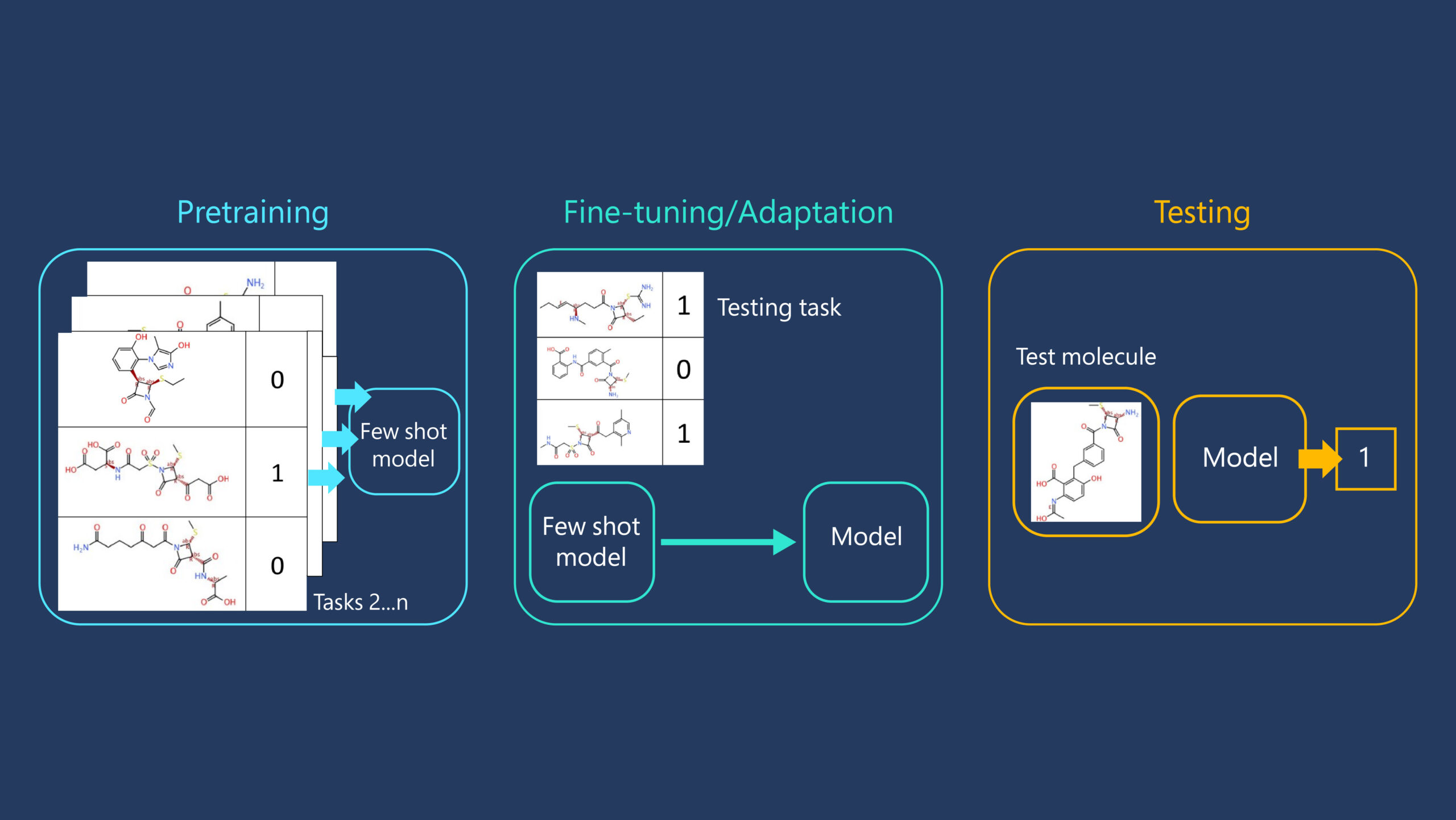
Several strategies for pretraining a few-shot learning model exist, and because the most effective approach to molecule-protein binding prediction given a limited amount of data is unclear, it is important to compare of a range of possibilities. To understand which method is most useful, the joint Microsoft Research and Novartis teams compared the following approaches.
Meta-learning approaches are trained with the objective of creating the fastest few-shot learner. For instance, model-agnostic meta-learning (opens in new tab) optimizes an objective that specifically measures how well a model adapts when it is specialised to a new task.
Prototypical networks (opens in new tab), another meta-learning method, predict the label of a new example by determining which examples in the support set are most similar.
Pretraining approaches aim to prepare an ML model for specialization by learning to identify the most relevant features. One such technique, multitask training simply aims to train a model to predict labels for molecules drawn from multiple tasks concurrently. In self-supervised pre-training, models are trained to recover removed or perturbed information in the input.
Spotlight: AI-POWERED EXPERIENCE

Microsoft research copilot experience
Discover more about research at Microsoft through our AI-powered experience
Building a curriculum
Comparing such methods is only fair if all few-shot learners are given the same testing task and had access to the same information during the pretraining phase. However, prior to our work, there was neither a well-defined set of tasks nor a clear testing strategy. Working with our collaborators at Novartis, we designed a dataset and testing procedure that reflects the real challenges in the early-stage drug-discovery process. We extracted data from ChEMBL (opens in new tab)—a publicly available database, performed in-depth cleaning and filtering on the data, and carefully assigned activity labels based on measured values.
A suitable pretraining curriculum must go together with rigorous testing, and we took great care to ensure that targets represented by pretraining were not repeated in our testing tasks. Here, we focused on testing tasks that represented the interaction of drug molecules with specific classes of enzymes so that overall results could also be broken down by performance on each class.
Making an assessment
How well do few-shot learning models perform in comparison to those ML models that are not given access to pretraining data? We need to understand the limits of model performance in the absence of pretraining to assess the real value in pretraining schemes.
To do this, we took a selection of pharmaceutical-industry standard models and supplied them with the support set data of our testing tasks, giving them the same treatment as the few-shot learning models at testing time. In fact, for both the pretrained few-shot methods and our untrained methods, we subjected them to testing across a range of tasks while supplying them with varying amounts of support set data.
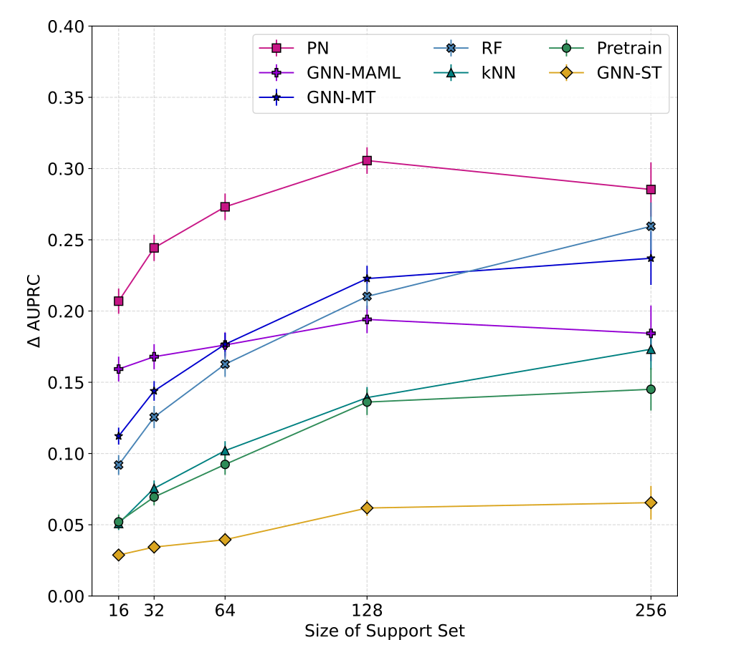
We found that even with no pretraining, models can do very well provided they have access to enough data during the test, but when they are given only a few datapoints, only models that have been pretrained can make good predictions. In Figure 3, we show a comparison of model performance on all testing tasks as more training data is supplied to each model. The results show an increase in performance over that of a completely uninformed classifier that randomly assigns a label to each new query molecule. While self-supervised pretraining and multitask approaches do not perform better than models that are not pretrained, the meta-learning approaches provided a significant boost. In particular, we demonstrated that prototypical networks are very effective in the setting of small amounts of data available in early-stage drug-discovery. This method had not previously been used, and we note that several promising improvements more tailored to molecular property prediction are possible in this setting.
Our work demonstrates that not only is early-stage drug-discovery well-posed as a few-shot learning problem, but also that pretraining and specifically meta-learning approaches can make valuable improvements to the quality of molecular property predictions. By publishing the dataset and evaluation framework, alongside these baseline results, we have enabled the drug-discovery community to access the latest state-of-the-art ML research on a truly realistic problem. At the same time, we aim to encourage ML researchers to address this challenging new domain.
By focusing ML research on early-stage drug-discovery, we aim to inspire new, well-suited modelling approaches and revolutionize the application of computation in pharmaceutical development. By reducing the necessity of synthesising and consequently in vitro testing of large numbers of molecules, these kinds of methods can help to reduce the time of taking a drug from initial concept to the market.



