

In 1928, Alexander Fleming accidentally let his petri dishes go moldy, a mistake that would lead to the breakthrough discovery of penicillin and save the lives of countless people. From these haphazard beginnings, the pharmaceutical industry has grown into one of the most technically advanced and valuable sectors, driven by incredible progress in chemistry and molecular biology. Nevertheless, a great deal of trial and error still exists in the drug discovery process. With an estimated space of 1060 small organic molecules (opens in new tab) that could be tried and tested, it is no surprise that finding useful compounds is difficult and that the process is full of costly dead ends and surprises.
The challenge of molecule design also lies at the heart of many applications outside pharmacology, including in the optimization of energy production, electronic displays, and plastics. Each of these fields has developed computational methods to search through molecular space and pinpoint useful leads that are followed up in the lab or in more detailed physical simulations. As a result, there are now vast libraries of molecules tagged with useful properties. The abundance of data has encouraged researchers to turn to data-driven approaches to reduce the degree of trial and error in chemical development, and the aim of our paper (opens in new tab) being presented at the 2018 Conference on Neural Information Processing Systems (NeurIPS) (opens in new tab) is to investigate how recent advances, specifically in deep learning techniques, could help harness these libraries for new molecular design tasks.
Deep learning with molecular data
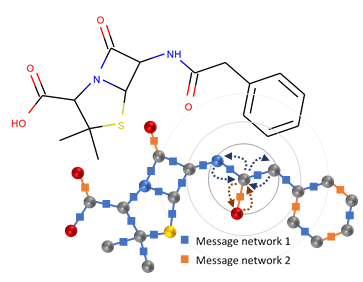
Figure 1: The chemical structure of naturally occurring penicillin (penicillin G) and its representation as a graph in a GGNN. The messages passed in the environment of a single node are shown as curved arrows, and the neural networks that transform the messages are shown as small squares. Repeated rounds of message passing allow each node to learn about its surroundings (gray circles).
Deep learning methods have revolutionized a range of applications requiring understanding or generation of unstructured data such as pictures, audio, and text from large datasets. Applying similar methods to organic molecules poses an interesting challenge because molecules contain a lot of structure that is not easy to concisely capture with flat text strings or images (although some schemes do exist (opens in new tab)). Instead, organic chemists typically represent molecules as a graph where nodes represent atoms and edges represent covalent bonds between atoms. Recently, a class of methods that have collectively become known as neural message passing has been developed precisely to handle the task of deep learning on graph-structured data. The idea of these methods is to encode the local information, such as which element of the periodic table a node represents, into a low-dimensional vector at each node and then pass these vectors along the edges of the graph to inform each node about its neighbors (see Figure 1). Each message is channeled through small neural networks that are trained to extract and combine information to update the destination node’s vector representation to be informative for the downstream task. The message passing can be iterated to allow each node to learn about its more distant neighbors in the graph. Microsoft Research developed one of the earliest variants of this class of deep learning models—the gated graph neural network (GGNN). Microsoft’s primary application focus for GGNNs is in the Deep Program Understanding project, where they are used to analyze program source code (which can also be represented using graphs). Exactly the same underlying techniques are applicable to molecular graphs.
Generating molecules
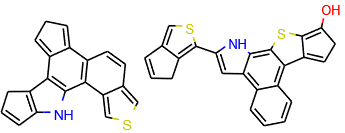
Figure 2: Example molecules generated by our system after being trained on organic solar cell molecules (CEP database (opens in new tab)).
Broadly speaking, there are two types of questions that a machine learning system could try to solve in molecule design tasks. First, there are discriminative questions of the following form: What is the property Y of molecule X? A system trained to answer such questions can be used to compare given molecules by predicting their properties from their graph structure. Second, there are generative questions—what is the structure of molecule X that has the optimum property Y?—that aim to invent structures that are similar to molecules seen during training but that optimize for some property. The new paper concentrates on the latter, generative question; GGNNs have already shown great promise in the discriminative setting (for example, see the code available here (opens in new tab)).
The basic idea of the generative model is to start with an unconnected set of atoms and some latent “specification” vector for the desired molecule and gradually build up molecules by asking a GGNN to inspect the partial graph at each construction step and decide where to add new bonds to grow a molecule satisfying the specification. The two key challenges in this process are ensuring the output of chemically stable molecules and designing useful low-dimensional specification vectors that can be decoded into molecules by the generative GGNN and are amenable to continuous optimization techniques for finding locally optimal molecules.
For the first challenge, there are many chemical rules that dictate whether a molecular structure is stable. The simplest are the valence rules, which dictate how many bonds an element can make in a molecule. For example, carbon atoms have a valency of four and oxygen a valency of two. Inferring these known rigid rules from data and learning to never violate them in the generative process is a waste of the neural network’s capacity. Instead, in the new work, we simply incorporate known rules into the model, leaving the network free to discover the softer trends and patterns in the data. This approach allows injection of domain expertise and is particularly important in applications where there is not enough data to spend on relearning existing knowledge. We believe that combining this domain knowledge and machine learning will produce the best methods in the future.
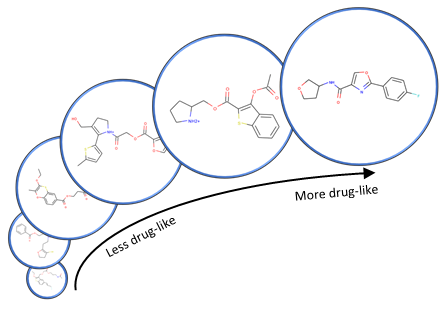
Figure 3: Example molecule optimization trajectory when optimizing the quantitative estimate of drug-likeness (QED) of a molecule after training on the ZINC database (opens in new tab). The initial molecule has a QED of 0.4, and the final molecule has a QED of 0.9
For the second challenge, we used an architecture known as a variational autoencoder to discover a space of meaningful specification vectors. In this architecture, a discriminative GGNN is used to predict some property Y of a molecule X, and the internal vector representations in this discriminative GGNN are used as the specification vector for a generative GGNN. Since these internal representations contain information about both the structure of molecule X and the property Y, continuous optimization methods can be used to find the representation that optimizes property Y; the representation is then decoded to find useful molecules. Example molecules generated by the new system are shown in Figures 2 and 3.
Collaborating with experts
The results in the paper are very promising on simple molecule design tasks. However, deep learning methods for molecule generation are still in their infancy, and real-world molecule design is a very complicated process with many different objectives to consider, such as molecule efficacy, specificity, side effects, and production costs. To make significant further progress will require collaboration of machine learning experts and expert chemists. One of the main aims of this paper is to showcase the basic capabilities of deep learning in this space and thereby act as a starting point for dialogue with chemistry experts to see how these methods could enhance their productivity and have the most impact.



